Query to reference mapping#
In this tutorial, we will demonstrate how CellMapper can be used in the general query-to-reference mapping scenario, where we have two datasets from the same modality (e.g. scRNA-seq). We’ll use scVI [LRC+18] to get a joint latent space, and we’ll use CellMapper in that space to transfer cell type labels, evaluate the quality of label transfer, and compute presence scores.
This tutorial is inspired by an scVI tutorial on the same dataset, and an HNOCA-tools tutorial [HDF+24] showcasing similar techniques.
Preliminaries#
Import packages & data#
%load_ext autoreload
%autoreload 2
import scanpy as sc
import numpy as np
import pandas as pd
from pathlib import Path
import cellmapper
import matplotlib.pyplot as plt
import torch
import scvi
import anndata as ad
sc.set_figure_params(scanpy=True, frameon=False, fontsize=10)
# some torch and scvi-tools settings
torch.set_float32_matmul_precision("high")
scvi.settings.dl_num_workers = 10 # will depend on your system
scvi.settings.dl_persistent_workers = True
MODEL_DIR = Path("models/lung")
ACCELERATOR = "mps" if torch.backends.mps.is_available() else "auto" # when runnign locally on Apple Silicon
Data loading#
Load the lung data [VBKB+19] that has been used in the scIB lung integration challenge [LButtnerC+22], following the scVI tutorial on Atlas-level integration of lung data.
adata = sc.read(
"data/lung_atlas_preprocessed.h5ad",
backup_url="https://figshare.com/ndownloader/files/52859312",
)
adata
AnnData object with n_obs × n_vars = 32472 × 2000
obs: 'dataset', 'location', 'nGene', 'nUMI', 'patientGroup', 'percent.mito', 'protocol', 'sanger_type', 'size_factors', 'sampling_method', 'batch', 'cell_type', 'donor'
var: 'highly_variable', 'highly_variable_rank', 'means', 'variances', 'variances_norm', 'highly_variable_nbatches'
uns: 'hvg'
layers: 'counts'
Prepare the data#
Compute a basic umap without batch correction.
sc.pp.pca(adata)
sc.pp.neighbors(adata)
sc.tl.umap(adata)
Split the data into query and reference datasets.
query_batches = ["4", "A2", "B3"]
adata.obs["split"] = pd.Categorical(
np.where(adata.obs["batch"].isin(query_batches), "query", "reference"), categories=["query", "reference"]
)
Visualize this split, some sample covariates, and cell type labels.
with plt.rc_context({"figure.figsize": (2.5, 2.5), "legend.fontsize": 6, "axes.titlesize": 8}):
sc.pl.embedding(
adata, basis="umap", color=["split", "location", "protocol", "batch", "cell_type"], ncols=3, wspace=0.3
)
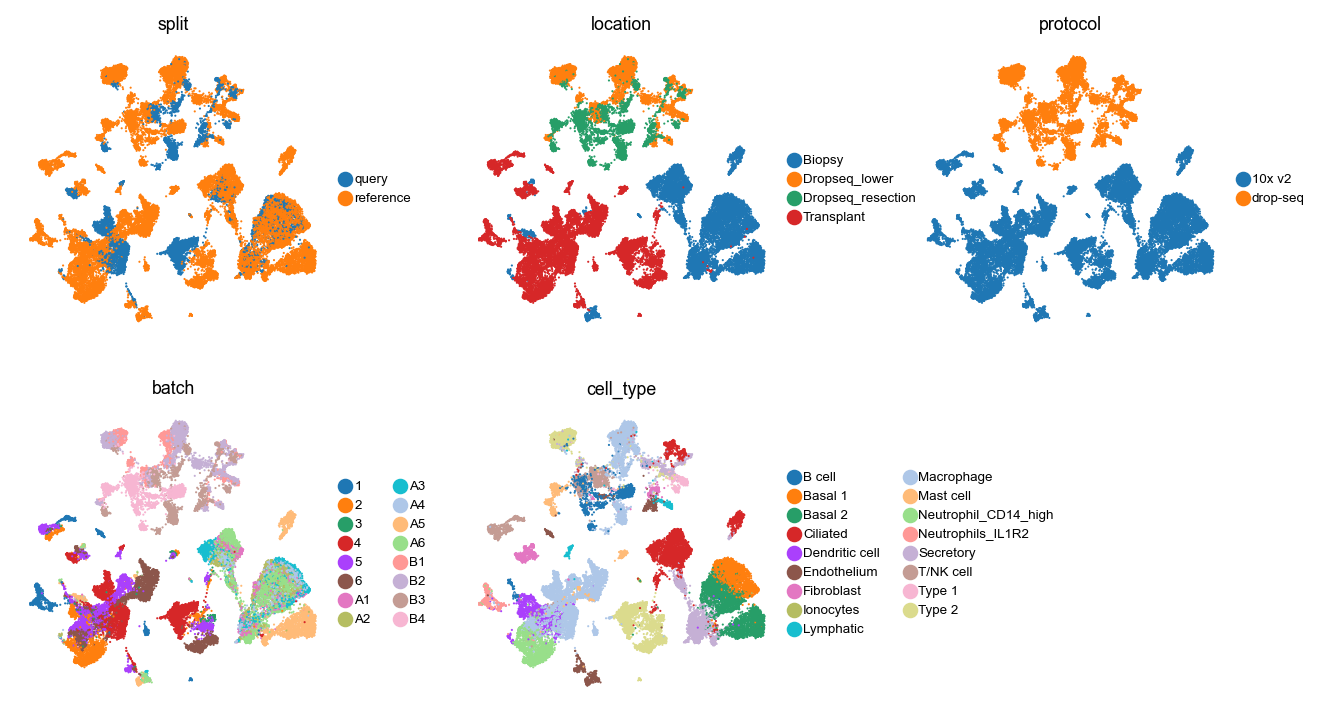
Split the AnnData into two objects.
ad_query = adata[adata.obs["split"] == "query"].copy()
ad_reference = adata[adata.obs["split"] == "reference"].copy()
Take a look at cell and gene numbers here.
ad_query.shape, ad_reference.shape
((7128, 2000), (25344, 2000))
Main analysis#
Train scVI reference model#
scvi.model.SCVI.setup_anndata(ad_reference, batch_key="batch", layer="counts")
accelerator = "mps" if torch.backends.mps.is_available() else "auto" # when runnign locally on Apple Silicon
Create and tain the reference model.
# scvi_ref = scvi.model.SCVI(
# ad_reference,
# use_layer_norm="both",
# use_batch_norm="none",
# encode_covariates=True,
# dropout_rate=0.2,
# gene_likelihood="nb",
# n_latent=30,
# n_layers=2,
# )
# scvi_ref.train(accelerator=ACCELERATOR)
# # write model to file
# scvi_ref.save(MODEL_DIR / "scvi_ref", overwrite=True)
Load a pre-trained model.
scvi_ref = scvi.model.SCVI.load(MODEL_DIR / "scvi_ref", adata=ad_reference, accelerator=ACCELERATOR)
INFO File models/lung/scvi_ref/model.pt already downloaded
Extract the latent space coordinates
SCVI_LATENT_KEY = "X_scVI"
ad_reference.obsm[SCVI_LATENT_KEY] = scvi_ref.get_latent_representation()
Update with the query#
scvi.model.SCVI.prepare_query_anndata(ad_query, scvi_ref)
INFO Found 100.0% reference vars in query data.
Create and train the query model
# scvi_query = scvi.model.SCVI.load_query_data(ad_query, scvi_ref)
# scvi_query.train(max_epochs=200, plan_kwargs={"weight_decay": 0.0}, accelerator=ACCELERATOR)
# # write model to file
# scvi_query.save(MODEL_DIR / "scvi_query", overwrite=True)
Load a pre-trained model.
scvi_query = scvi.model.SCVI.load(MODEL_DIR / "scvi_query", adata=ad_query)
INFO File models/lung/scvi_query/model.pt already downloaded
Extract the latent space coordinates
ad_query.obsm[SCVI_LATENT_KEY] = scvi_query.get_latent_representation()
Visualize query and reference data jointly#
Concatenate query and reference objects and compute a UMAP in the shared latent space.
ad_combined = ad.concat([ad_query, ad_reference])
ad_combined
AnnData object with n_obs × n_vars = 32472 × 2000
obs: 'dataset', 'location', 'nGene', 'nUMI', 'patientGroup', 'percent.mito', 'protocol', 'sanger_type', 'size_factors', 'sampling_method', 'batch', 'cell_type', 'donor', 'split', '_scvi_batch', '_scvi_labels'
obsm: 'X_pca', 'X_umap', 'X_scVI'
layers: 'counts'
Let’s get the latent space coordinates
SCVI_UMAP_KEY = "X_umap_scVI"
sc.pp.neighbors(ad_combined, use_rep=SCVI_LATENT_KEY)
sc.tl.umap(ad_combined, key_added=SCVI_UMAP_KEY)
Take a look at this in the umap.
with plt.rc_context({"figure.figsize": (2.5, 2.5), "legend.fontsize": 8, "axes.titlesize": 8}):
sc.pl.embedding(ad_combined, basis=SCVI_UMAP_KEY, color=["batch", "split", "cell_type"], wspace=0.4)
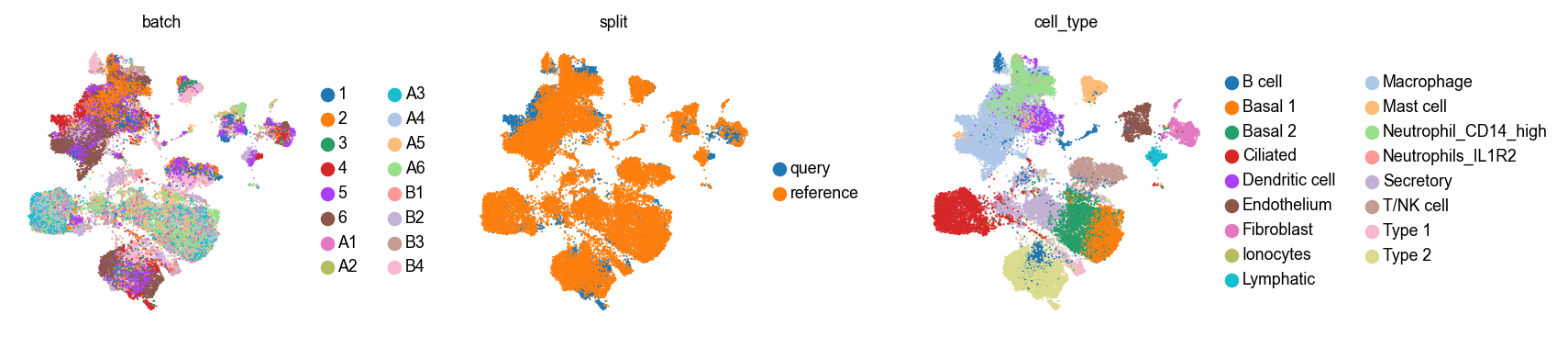
Copy this joint umap back into the individual AnnData objects.
ad_query.obsm[SCVI_UMAP_KEY] = ad_combined[ad_combined.obs["split"] == "query"].obsm[SCVI_UMAP_KEY].copy()
ad_reference.obsm[SCVI_UMAP_KEY] = ad_combined[ad_combined.obs["split"] == "reference"].obsm[SCVI_UMAP_KEY].copy()
Work with CellMaper#
Transfer celltype labels#
We’ll use a non-default mapping method below (hnoca). You can experiment a bit with this - CellMapper implements many different ways of turning k-NN graphs into mapping matrices. hnoca is a certain normalization of Jaccard similarities, following the original implementation in HNOCA-tools.
cmap = cellmapper.CellMapper(ad_query, ad_reference).map(
obs_keys=["cell_type"], use_rep=SCVI_LATENT_KEY, kernel_method="hnoca"
)
cmap
INFO Initialized CellMapper with 7128 query cells and 25344 reference cells.
INFO Using sklearn to compute 30 neighbors.
INFO Computing mapping matrix using kernel method 'hnoca'.
INFO Mapping categorical data for key 'cell_type' using direct multiplication.
INFO Categorical data mapped and stored in query.obs['cell_type_pred'].
CellMapper(query=AnnData(n_obs=7,128, n_vars=2,000), reference=AnnData(n_obs=25,344, n_vars=2,000)
Let’s compare the predicted labels with ground truth
cmap.evaluate_label_transfer("cell_type")
INFO Accuracy: 0.7779, Precision: 0.9291, Recall: 0.7779, Weighted F1-Score: 0.8241, Macro F1-Score: 0.7221,
Excluded Fraction: 0.0000
A weighted F1 score of 0.82 is quite good here. Let’s take a look at the confusion matrix to learn more.
cmap.plot_confusion_matrix("cell_type", normalize="true", include_values=False, figsize=(8, 7))
<Axes: title={'center': 'Confusion Matrix'}, xlabel='Predicted label', ylabel='True label'>
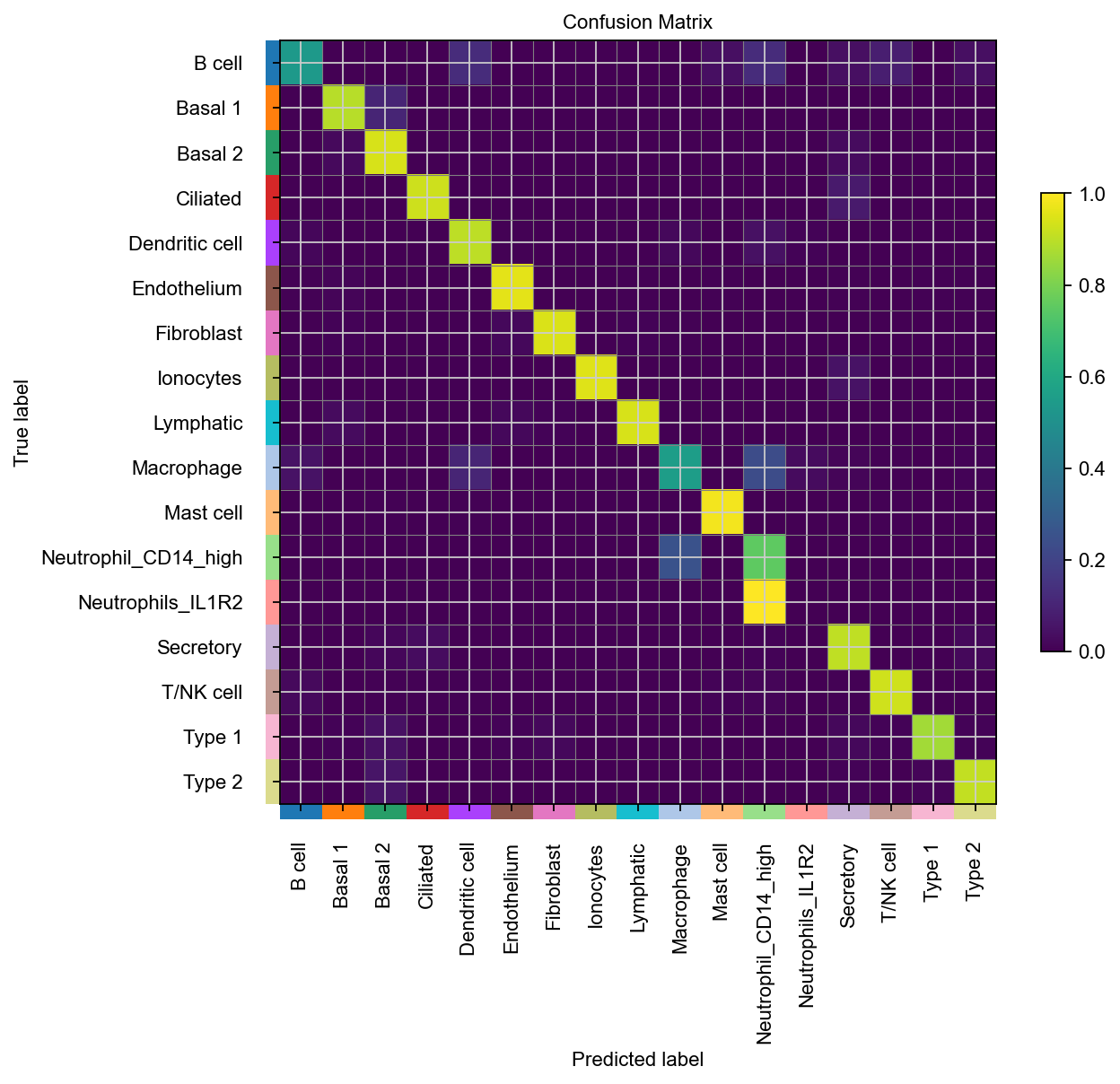
For most cell types, we’re doing quite well. Let’s also compare ground-truth with predicted labels in the umap.
with plt.rc_context({"figure.figsize": (2.5, 2.5), "legend.fontsize": 6, "axes.titlesize": 8}):
sc.pl.embedding(ad_query, basis=SCVI_UMAP_KEY, color=["cell_type", "cell_type_pred", "cell_type_conf"], wspace=0.7)
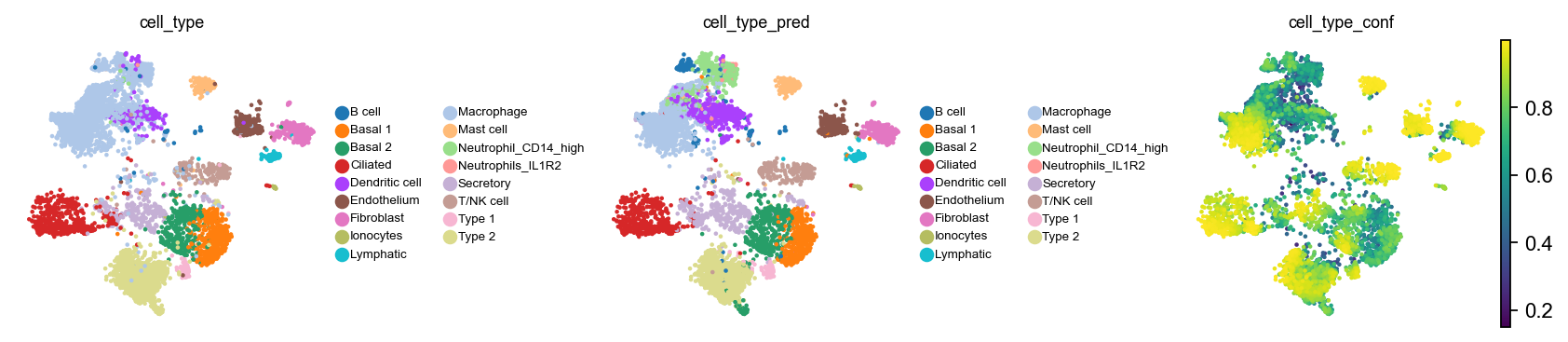
For cells where the prediction does not match the ground truth label, it looks like the confidence score is usually lower.
Compute presence scores#
Next, we would like to figure out how well represented each query cell is in the reference atlas. Such an analysis is particularly useful in disease studies, where particular cell types might be depleted or enriched, compared to a healthy reference atlas.
cmap.compute_presence_score(groupby="batch")
INFO Presence score across all query cells computed and stored in `reference.obs['presence_score']`
INFO Presence scores per group defined in `query.obs['batch']` computed and stored in
`reference.obsm['presence_score']`
Following the corresponding implementation in HNOCA-tools [HDF+24], we smooth out the presence scores a bit.
smap = cellmapper.CellMapper(ad_reference).map(obs_keys="presence_score", t=5, use_rep=SCVI_LATENT_KEY)
INFO Initialized CellMapper for self-mapping with 25344 cells.
INFO Self-mapping mode detected. Computing only yx neighbors for efficiency (all neighbor matrices will contain
the same information).
INFO Using sklearn to compute 30 neighbors.
INFO Computing mapping matrix using kernel method 'umap'.
INFO Mapping numerical data for key 'presence_score' with t=5 steps using iterative diffusion_method.
INFO Numerical data mapped and stored in query.obs['presence_score_pred'].
Visualize these scores on the joint umap.
with plt.rc_context({"figure.figsize": (2.5, 2.5), "legend.fontsize": 8, "axes.titlesize": 8}):
sc.pl.embedding(ad_reference, basis=SCVI_UMAP_KEY, color=["batch", "presence_score_pred", "cell_type"], vmax="p99")
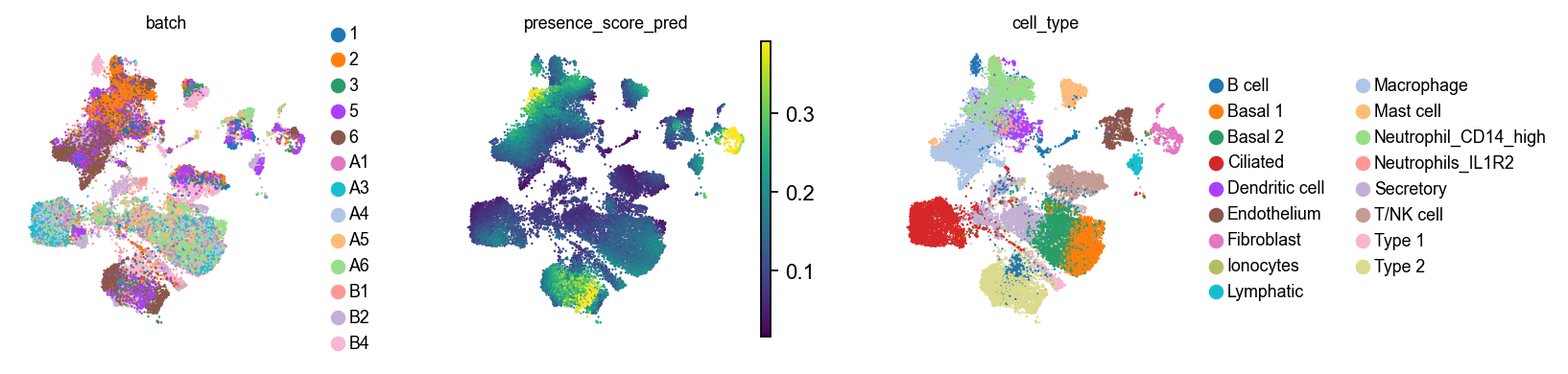
We can now stratify this by cell type in the reference.
sc.pl.violin(ad_reference, keys="presence_score_pred", groupby="cell_type", rotation=90)
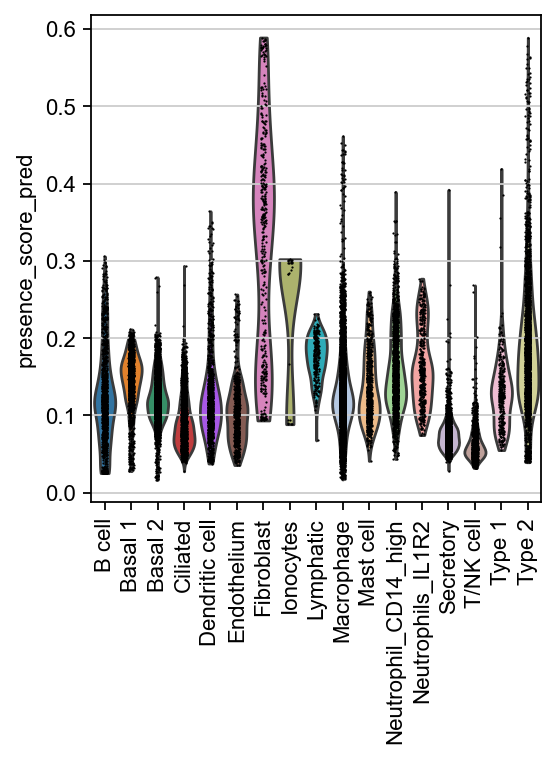
It looks like our query dataset is enriched for Fibroblasts, relative to the reference. However, the query itself consitst of three batches - let’s check whether these batches are equally represented in the reference, or whether there are differences. For this, CellMapper wrote a DataFrame to .obsm - we also apply some smoothing to that here.
smap = cellmapper.CellMapper(ad_reference).map(obsm_keys="presence_score", t=5, use_rep=SCVI_LATENT_KEY)
INFO Initialized CellMapper for self-mapping with 25344 cells.
INFO Self-mapping mode detected. Computing only yx neighbors for efficiency (all neighbor matrices will contain
the same information).
INFO Using sklearn to compute 30 neighbors.
INFO Computing mapping matrix using kernel method 'umap'.
INFO Mapping embeddings for key 'presence_score' with t=5 steps using iterative diffusion_method
INFO Embeddings mapped and stored in query.obsm['presence_score_pred']
Let’s copy the final presence scores per batch into .obs and visualize.
obsm_key = "presence_score_pred"
obs_score_names = [f"{obsm_key}_{key}" for key in query_batches]
for obsm_name, obs_name in zip(query_batches, obs_score_names, strict=False):
ad_reference.obs[obs_name] = ad_reference.obsm[obsm_key][obsm_name]
Visualize in the UMAP
with plt.rc_context({"figure.figsize": (2.5, 2.5), "legend.fontsize": 6, "axes.titlesize": 8}):
sc.pl.embedding(ad_reference, basis="umap_scVI", color=obs_score_names + ["cell_type"], vmax="p99", ncols=3)
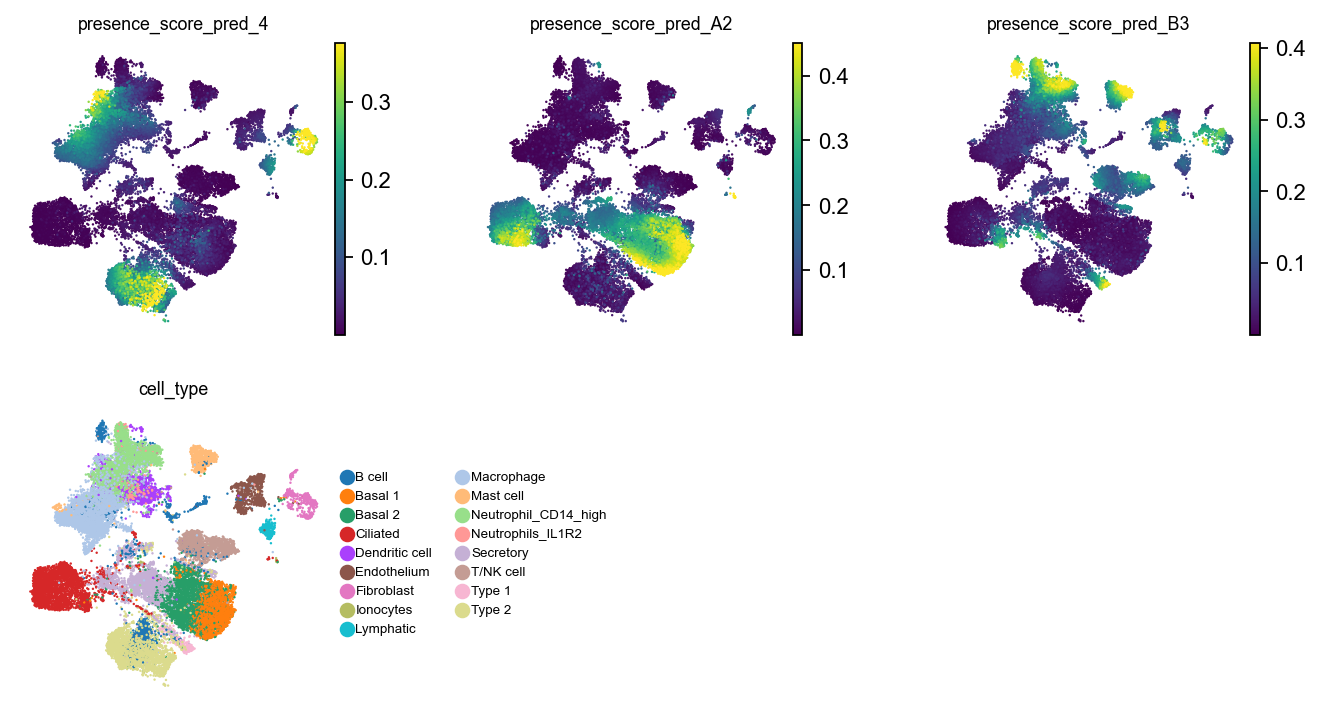
It looks like the different query batches cover different parts of the phenotypic manifold described by the reference dataset. We can summarize this result in a dotplot.
# Set up dummy AnnData object
X = ad_reference.obs[obs_score_names].values
dummy_adata = ad.AnnData(
X=X,
obs=ad_reference.obs[["cell_type"]].copy(),
var=pd.DataFrame(index=obs_score_names), # features as .var_names
)
sc.pl.dotplot(dummy_adata, var_names=obs_score_names, groupby="cell_type", color_map="viridis", swap_axes=True)
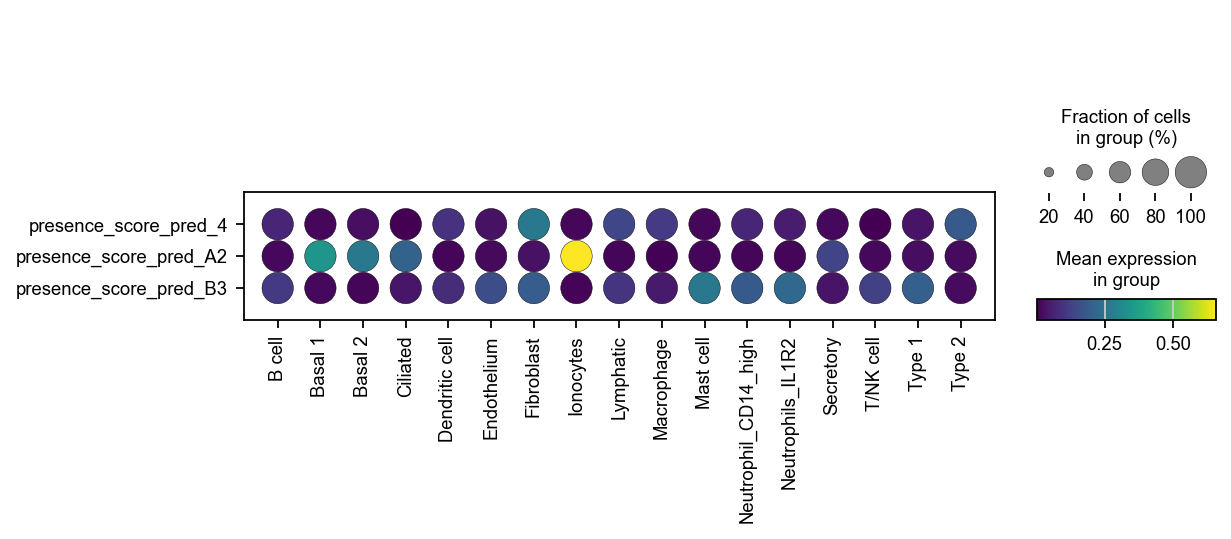
We now see that the enrichment for fibroblasts stems only from a single batch - the other query batches cover different parts of the reference landscape.